Computational Materials Group (CMG) lead by Prof. D. Aksyonov is developing new electrode materials for metal-ion batteries using computational methods. The supreme ingredient of success is a deep understanding of the material’ properties on the atomic and electronic level, which can be achieved with modeling.
Main directions:
- Computational design of solid/solid and solid/liquid interfaces for metal-ion batteries (Applied Surface Science 537 (2021) 147750)
- Advanced study of defects in electrodes for metal-ion batteries (Inorg. Chem. 2021, 60, 5497−5506)
- Understanding cation migration barriers in oxide and phosphate based cathode materials with DFT calculations (Computational Materials Science 154 (2018) 449–458)
- Development of computational framework SIMAN for high-throughput DFT calculations (https://github.com/dimonaks/siman)
- To advanced high capacity layered electrode materials for lithium-ion batteries through the understanding of oxidation-reduction processes at the atomic level
- Atomic-level Understanding of Interface Structure Evolution and Engineering Guidelines for Next Li-ion Solid State Batteries
- Search for new materials for gas electrodes of lithium- and sodium-oxygen current sources: predictive computer simulation and experimental testing

Figure 1. Easier formation of surface antisite defect in layered oxides discovered with DFT+U oxides. From Applied Surface Science 537 (2021) 147750

Figure 2. Combined DFT + X-Ray + Neutron diffraction refined hydrogen defects in LiFePO4 cathode material. From Inorg. Chem. 2021, 60, 5497−5506

Figure 3. Li diffusion pathways for three classes of crystal structures: (a) spinel, (b) olivine, and (c) tavorite. From Computational Materials Science 154 (2018) 449–458
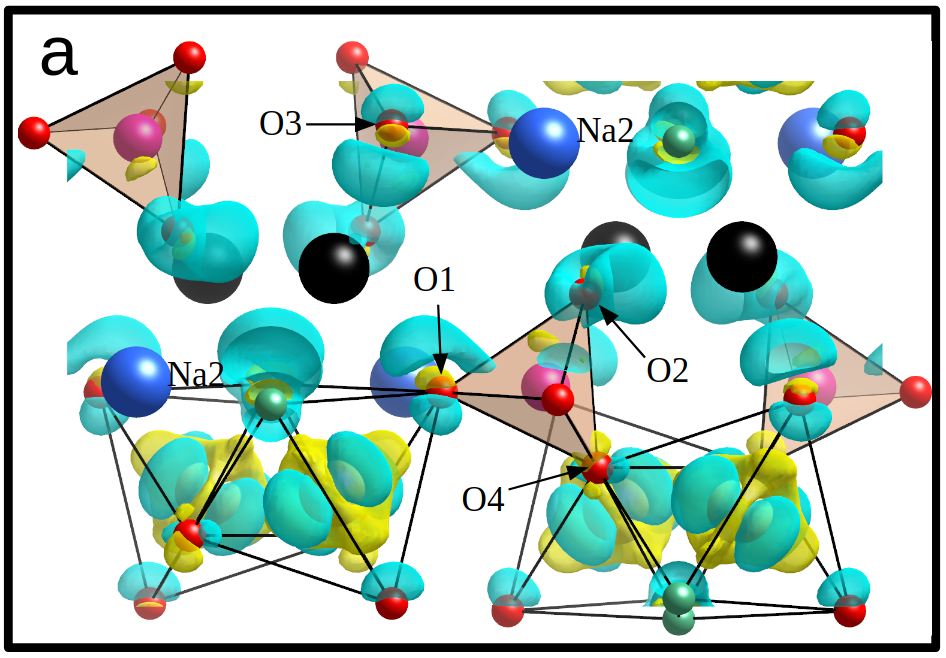
Figure 4. Change of charge density in Na2FePO4 after extraction of sodium (black spheres). From J. Am. Chem. Soc. 2018, 140, 3994−4003.